Structure and dynamics of water and ions in the nanometer channel of elemental doped ettringites
Ocean resources exploration and utilization have attracted extensive attention in industry and academia due to the increasingly acute shortage of fossil fuels [1]. Along with the construction of offshore infrastructure (e.g., wind power facilities, tidal power stations, oil platforms, and so on), ocean development has expanded from offshore and shallow-sea areas into deep-sea regions (ocean depth < 200 m) [2–4]. Cementitious materials play an extraordinarily important role in infrastructure construction by binding aggregates and providing concrete with strength [5]. The durability of concrete in ocean environments can differ greatly from that in routine natural environments, especially due to higher hydraulic pressure, higher temperatures, and chemical interactions with various sea ions [6]. The ocean's hydraulic pressure would increase by about 0.1 MPa for every 10 m of depth increase [7]. The ocean's mean temperature is about 3.5°C, and it ranges from 0 to 6°C in the deep-sea environment (at 3000 m). The common sea ions, including chloride, sulfate, and carbonate ions, attract researchers’ attention due to their relatively high corrosiveness [8, 9]. Moreover, the influence of chloride salts on concrete durability and performance has been demonstrated, and numerous methods have been proposed to delay concrete corrosion [10, 11]. Two main measures to reduce the influence of chloride ions are to reduce their concentration and improve resistance to chloride-ion penetration [12, 13]. In detail, on the one hand, source control is proposed to avoid the induction of chloride ions in concrete by chloride salt contained in mineral admixtures or water. On the other hand, significant efforts have been made to adjust concrete microstructures to control chloride-ion penetration [14]. To achieve this purpose, many theoretical and experimental studies have been conducted to illustrate the transport behavior of water or ions in concrete [15, 16]. Interestingly, because of the complexity of concrete pore structures, we need to account for the transport and penetration of chloride ions at different spatial scales. Many nanopores or nanochannels form during the hydration of calcium silicate hydrate (C-S-H), a typical binding phase in cement paste [17]. These nano-scaled pores or spaces would act as the channels for water or ion transport. Due to the apparent difference between these ultraconfined channels and the micro- or macro-pores, the transport behavior of water or ions in these nano-scaled channels is quite different. Besides, the cementitious concretes used in offshore or deep-sea infrastructures would withstand the influence of the continuous hydraulic pressure, low temperatures, and specific sea ions’ erosions during long-term service [18–20]. Thus, investigating the interactions between water or seawater ions and marine concretes under hydraulic pressure and low-temperature coupling conditions helps us understand the structural and chemical variations in these materials. Although the penetration and transport behavior of water or ions in concrete has been widely studied, the combined effect of high hydraulic pressure and low temperature in the deep-sea environment on the transport of water and ions through ultra-confined concrete channels remains poorly understood and systematically investigated.
For concrete, the hydration of the cementitious materials is absolutely central to achieving binding or strength [21]. Thus, the structural and component stability of hydration products (Calcium silicate hydrate (C-S-H), ettringite, portlandite (Ca(OH)2, CH), calcium-aluminate-silicate-hydrate (C-A-S-H), and AFm (short for a family of hydrated calcium aluminate phases)) of cementitious materials is crucial to guarantee the durability and stability of cementitious materials-based infrastructures [22–25]. Among them, ettringite can also be very important to the performance of marine concrete, along with C-S-H. It is the second main component of the hydration products of sulfoaluminate cement and is regarded as the volume-unstable factor of Portland cement [26]. Thus, investigating the influence of structural parameters on ettringite stability can be valuable for understanding its service performance and for marine concrete design [27]. To date, the effects of various elemental dopants on ettringite stability have been widely explored, yielding meaningful results. Interestingly, it has been reported that iron doping can reduce the stability of ettringite [28–32]. In addition, iodine species exhibit a strong affinity for ettringite [33]. Molecular dynamics (MD) simulation is a convenient method for evaluating the effects of elemental doping on ettringites in solutions with different ionic concentrations [34–37]. But the effect of hydraulic pressure on these interaction systems has seldom been explored.
In this work, atomistic models of the pristine and elemental (Sr2+, Fe3+, and IO3−) doped ettringites were constructed separately to study the transport behavior of water and Na+ or Cl− ions in the nanopore. In detail, different hydraulic pressures corresponding to different ocean depths are applied to the different ettringite models throughout the molecular dynamics simulation. The statistical and dynamic distributions of water and ions (Na+ or Cl−) in the pristine or elemental doped ettringite nanopore have been systematically explored by the density distribution analysis, radial distribution functions (RDF) analysis, time correlation function (TCF) analysis, and mean square displacement (MSD) analysis [38]. A plausible nanoscale explanation is proposed for how ocean depth and the ettringite component affected the transport of water and ions through the ettringite nanopore. The findings provide a materials science basis for the pristine and doped ettringites in deep-sea environments, which can be important for exploring deep-sea concrete bottom-up with expected performance and durability.
2 Theoretical and computational details
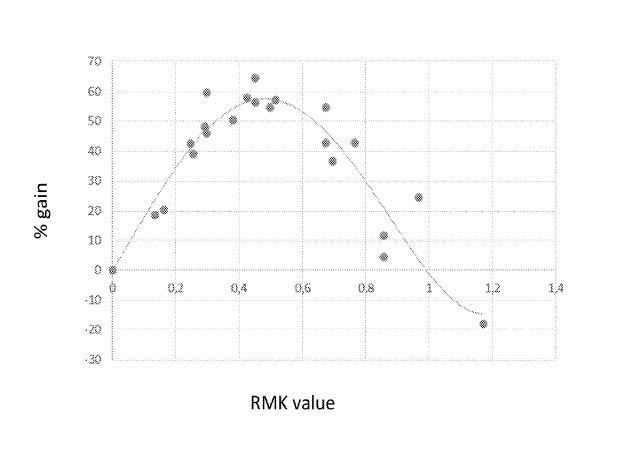
Based on the crystalline structure of pristine ettringite, the supercell is created by cleaving the crystal along the (001) plane [39]. Then, a 5.5 nm wide channel was created by separating the upper and lower ettringite (001) substrates. An ettringite nanopore model is built with the following parameters: a = 44.92 Å, b = 38.90 Å, c = 97.34 Å, and α = β = γ = 90°. The NaCl solution is used to fill the ettringite nanopore by randomly placing Na+, Cl−, and H2O molecules at the center of the 5.5 nm channel to achieve a 0.6 mol/L NaCl environment. For the Sr2+, Fe3+, and IO3− doped ettringite systems, the Ca2+, Al3+, or SO42− ions around the inner wall of the ettringite nanopore are replaced, respectively. Four doping degrees (2%, 10%, 15% and 20%) are investigated in our study (See Figure 1). At the very beginning, all of the ions were placed far away (approximately 20 Å) from the ettringite substrate to minimize the influence. The adsorption behavior of the ions was then explored by studying their structural and dynamic behaviors near the surface during MD simulations.
Figure 1 Ball and stick model of aqueous NaCl/ettringite interaction systems
The time step of 1 fs was set to ensure the accuracy of the simulation. A relatively long MD simulation (about 50 ns) was carried out to guarantee the full equilibrium of the simulated systems under the NPT ensemble. Then another MD calculation (200 ps) was applied to collect the MD trajectory. The Verlet algorithm was used for trajectory integration. The equilibrium trajectory of all atoms in the model was output every 1 ps. Finally, the balanced dynamic trajectory of all atoms was analyzed to explore the interaction of different ettringites with water and ions.
2.2 Force fields and MD procedures
ClayFF force fields, which are widely used to investigate hydrated and multi-component minerals, including cementitious materials/aqueous interaction systems, are utilized to carry out the whole MD simulations [39–42]. The atomistic interactions in ClayFF force fields are mainly composed of the Coulomb force, the Van der Waals force, the energy of bond stretching, and bond angle bending, as shown in Equations (1)–(5) [43].
The MD simulations in this study were carried out with the open-source software GROMACS. In detail, all the computation process involves three steps: (1) energy minimization, the simulation systems are fully relaxed, (2) pre-equilibrate, the simulation systems achieve to an equilibrate state through a relative long-term MD calculation (5 ns) under canonical ensemble (NPT), (3) MD calculation, the energy and atomistic spatial position variations relate to time are recorded for the following detailed analysis. During the MD calculations, the hydrostatic pressure of the systems is set according to the five sea depths (0, 3000, 5000, 10,000, and 20,000 m), considering the realistic deep-sea environment.
Finally, the equilibrated dynamic trajectory of all atoms in the interaction systems was analyzed to explore the ettringite/aqueous solution interactions, considering the impact of the elemental doping and the deep-sea environments.
3.1 The local structure of Na and Cl ions in pristine or elemental doped Ettringite’s nano-chanells
The density profile of the Na+ or Cl− in pristine ettringite’s nanochannels could reflect the interaction strength between the ions and ettringites. According to Figure 2, Na+ tends to accumulate around 2–2.3 Å from the ettringite (001) surface, while Cl− tends to accumulate around 2.3–2.7 Å from the ettringite (001) surface. Thus, Na+ exhibits a stronger affinity for the ettringite surface than Cl−. The difference in ionic radii between Na+ and Cl− might cause this phenomenon. The smaller Na+ exhibits better mobility.
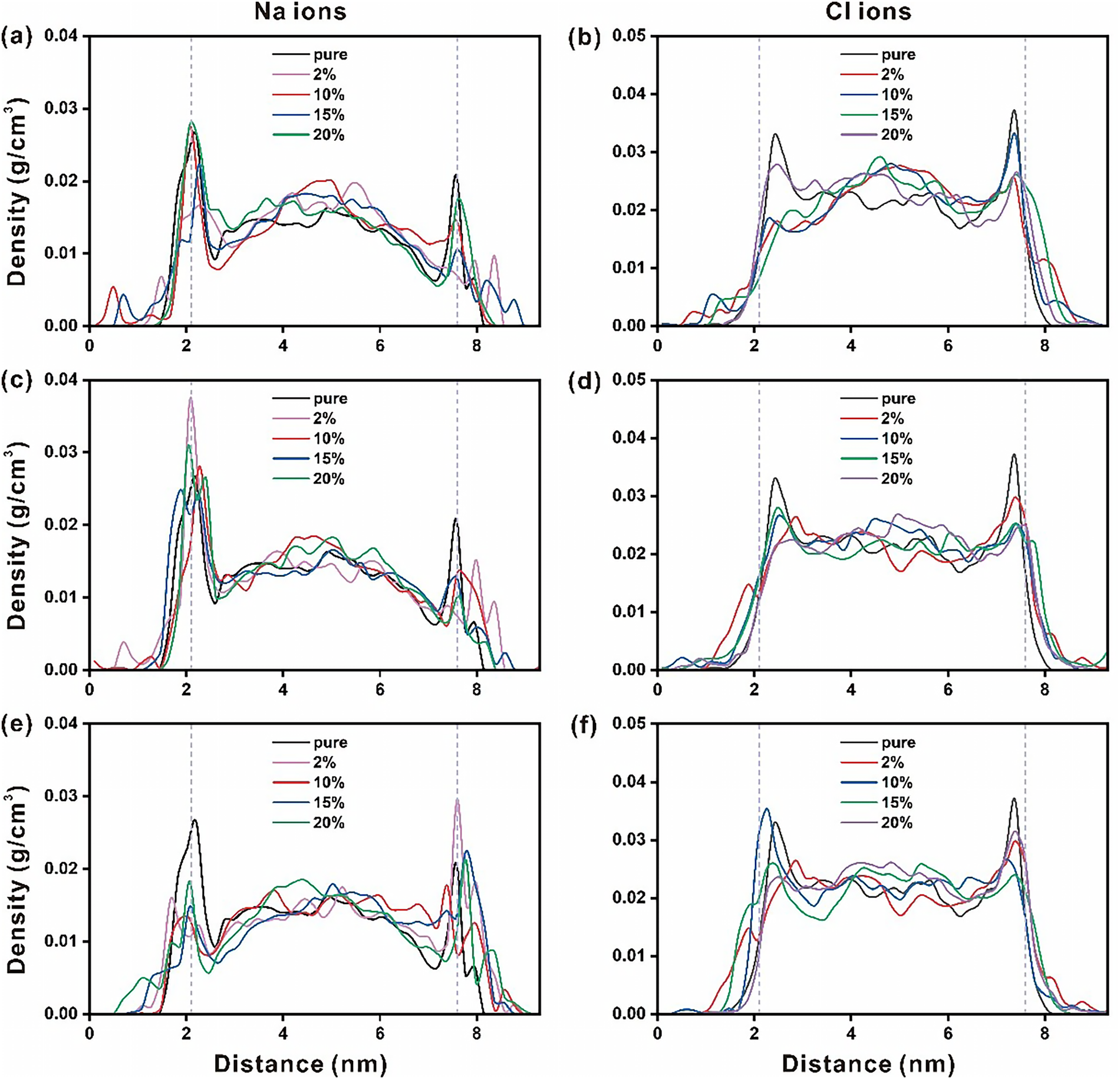
Figure 2 Density profiles for Na+ and Cl− in the nanopore of Sr2+ doped ettringite (a,b), Fe3+ doped ettringite (c,d) and IO3− doped ettringite (e,f)
Besides, the influence of the elemental doping on the ions/ettringites interactions was also analyzed. For the Sr2+ doped ettringite, an interesting phenomenon is that more Na+ and Cl− ions appear at the locations closer to the ettringite (001) surface. Based on our previous studies [44], the volume of the Ca-O polyhedron could be increased by Sr doping. The ionic radius of Sr is almost 30% larger than that of Ca. Thus, a very tiny extent change of the ettringite (001) surface structure caused by Sr doping strengthened the Na+ or Cl–/ettringite interactions. For the Fe-doped ettringite, similar results can be found that Na+ exhibits better affinity to ettringite compared to Cl–. Also, Fe doping could enhance the ions/ettringites interactions to some extent. This can also be illustrated by the difference in the ionic radius between Fe and Ca. A similar phenomenon is also observed in IO3– doped ettringite/ions interaction systems [45]. Both cation and anion doping can lead to changes in the surface structure of perovskite, thus affecting ion adsorption on the perovskite surface. Doping degree affects the ion distributions to some extent for both Sr, Fe, and IO3− doping. Besides, the competitive adsorption and blocking effects of Na+, Cl−, and other ions can be another factor to influence the ion distributions [46–49].
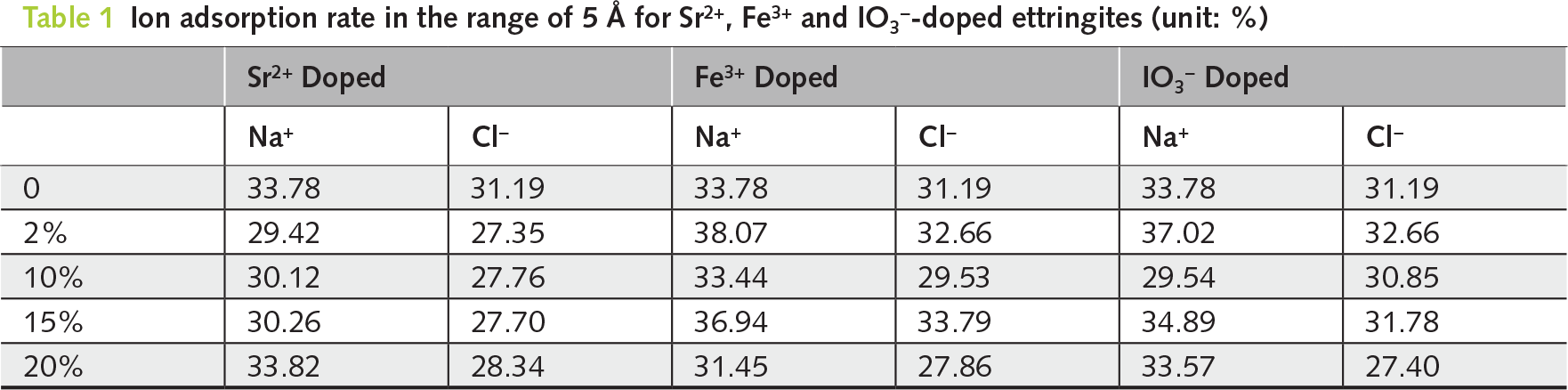
The Na+ and Cl− distributions in pristine or elementally doped ettringites were further analyzed using ion adsorption rate calculations (see Table 1). For Sr2+ doped ettringite, the ion adsorption rate of Na+ is larger than that of Cl−. It corresponds well with the results of the density profile. Besides the ionic radius difference of Na and Cl, the Sr-doped ettringite (001) surface state is another reason. Widely distributed O atoms on the very top of different ettringites’ surfaces exhibit relatively strong interactions with Na+. Further, the doping degree has little influence on the ion adsorption rates. The Fe3+ or IO3− doped ettringites also exhibit similar results.
The radial distribution function (RDF) analysis is another useful tool to explore the ion interactions [50]. Here, the RDF of Na-Cl pair, Na-O (O of hydroxyl groups on ettringites) pair and Ca-Cl pair on pristine or Sr2+-, Fe3+- and IO3−-doped ettringites with different doping degrees are analyzed and shown in Figure 3. It indicated that three peaks appear around 3 Å, 5.2 Å and 7.4 Å for Na-Cl pair corresponding to Na-Cl ionic bonding interactions, hydrated sodium ion-Cl interactions and ion cluster interactions (Na-O of water-Ca-Cl) (see Figure 3a). Among the three Na-Cl interaction types (Figure 3d, g), the ion cluster model exhibits the weakest interaction due to the relatively long distance between the Na and Cl ions. The doping degree will not change the interaction style between Na+ and Cl−.
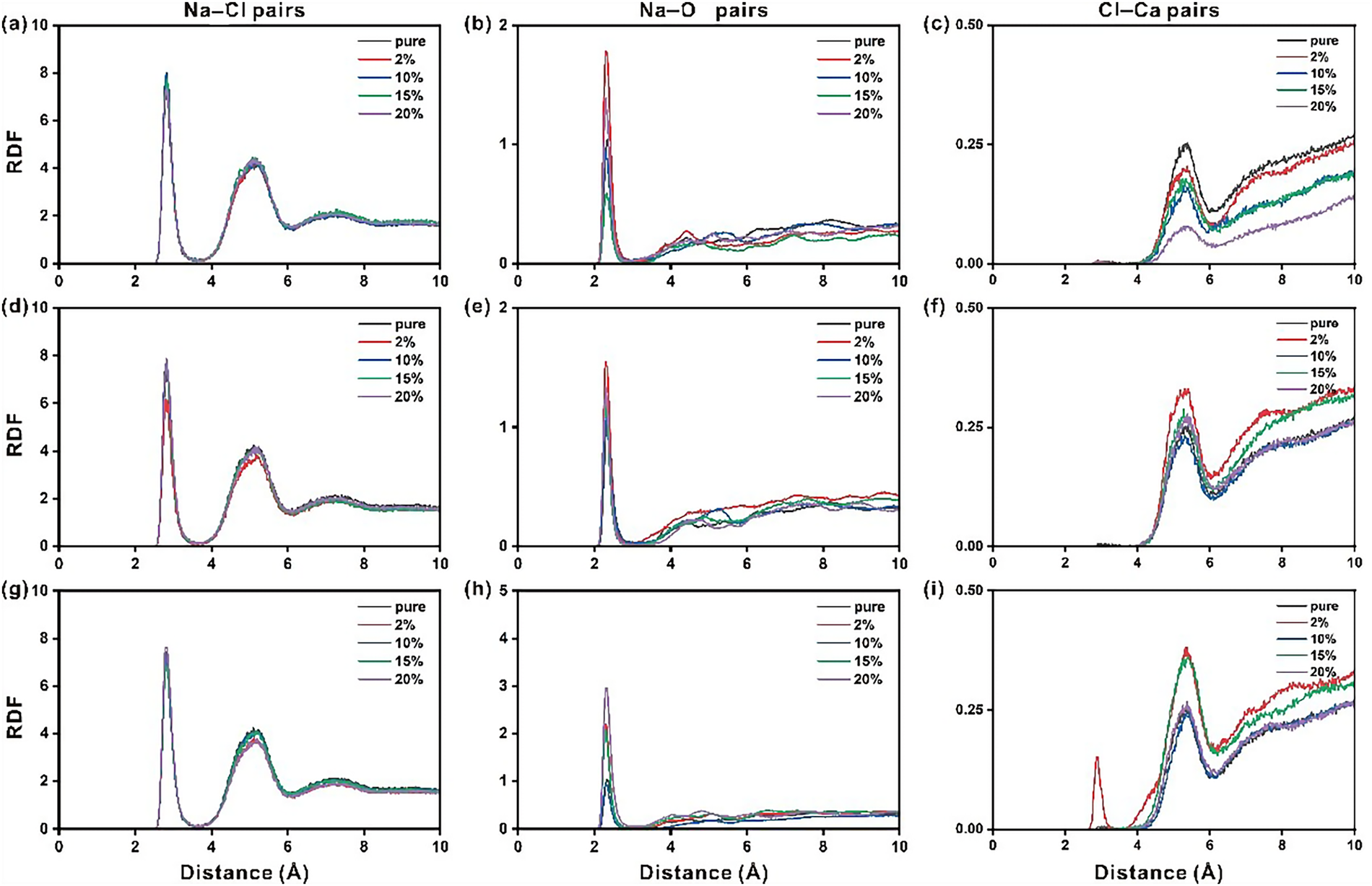
Figure 3 Radial distribution function of Na-Cl, Na-O and Cl-Ca pairs on the nanopore of Sr2+ doped ettringite (a–c), Fe3+ doped ettringite (d–f) and IO3− doped ettringite (g–i)
For the Na-O pair, the apparent peaks can be found around 2.4 Å, which refers to the Na-O bond length, and it corresponds well to the experimental measurements (2.3 to 2.5 Å). Besides, several relatively weak peaks exist after 3 Å, which refer to the weak interaction type between Na ions and O of hydroxyl groups on doped ettringite through the bridging role of the O atom of water. Interestingly, elemental doping affects the interactions between Na ions and O of hydroxyl groups on doped ettringite. For the Sr2+ doped ettringite (see Figure 3b), 2% and 20% doping enhanced the Na-O interactions, while 10% and 15% doping reduced the Na-O interactions. For the Fe3+ doped ettringite (see Figure 3e), no apparent changes can be found. For the IO3− doped ettringite (see Figure 3h), the Na-O interactions can be enhanced by IO3− doping. Nevertheless, it is reasonable to conclude that elemental doping could regulate the Na-O interactions by changing the global configurations of ettringites and changing the configurations of surficial hydroxyl groups in turn.
For the Cl-Ca pair, the apparent peaks appear around 5.4 Å, which refers to the interactions between Cl ions and the Ca ion of ettringites through the bridging of water molecules of the hydration layer. Different doping elements exhibit different regulatory behavior on Cl-Ca interactions. For Sr2+ doped ettringite, Cl-Ca interactions are reduced with the increase of the doping degree (Figure 3c). For Fe3+ doped ettringite, it is concluded that Cl-Ca interactions could be regulated by doping degree (Figure 3f). It is worth noting that the strong Cl-Ca interactions are found around 2.8 Å with 2% IO3− doping (Figure 3i). It is very close to the atomistic distance between Ca and Cl in CaCl2.
3.2 Dynamic properties of ions in nanopores
Time correlation function (TCF) analysis is carried out to further explore the stability of ionic interactions [51]. The TCF results of ionic pairs of Na-Cl, Na-Oh, and Cl-Ca on Sr2+, Fe3+, or IO3− doped ettringite are collected and shown in Figure 4. For Sr2+ doped ettringite, comparing with the TCF of Na-Cl pairs (see Figure 4a–c), the relatively small slope of the TCF of Na-Oh pairs indicates the quite stable ionic interactions between Na+ and O of alumina octahedrons of different doped ettringite. Besides, the doping degree (2% to 20%) exhibits little influence on the stability of ionic interactions for both Na-Cl and Na-Oh pairs. For the Cl-Ca pairs, the stability of Cl-Ca pairs is better than that of Na-Cl pairs, and similar to that of Na-Oh pairs. The doping degree influence on the TCF of Cl-Ca pairs is relatively complex. It is interesting that with 10% Sr-doped ettringites exhibit improved Cl-Ca interactions. At the same time, the Cl-Ca interactions can be weakened to some extent under the other doping degrees (2%, 15%, and 20%).
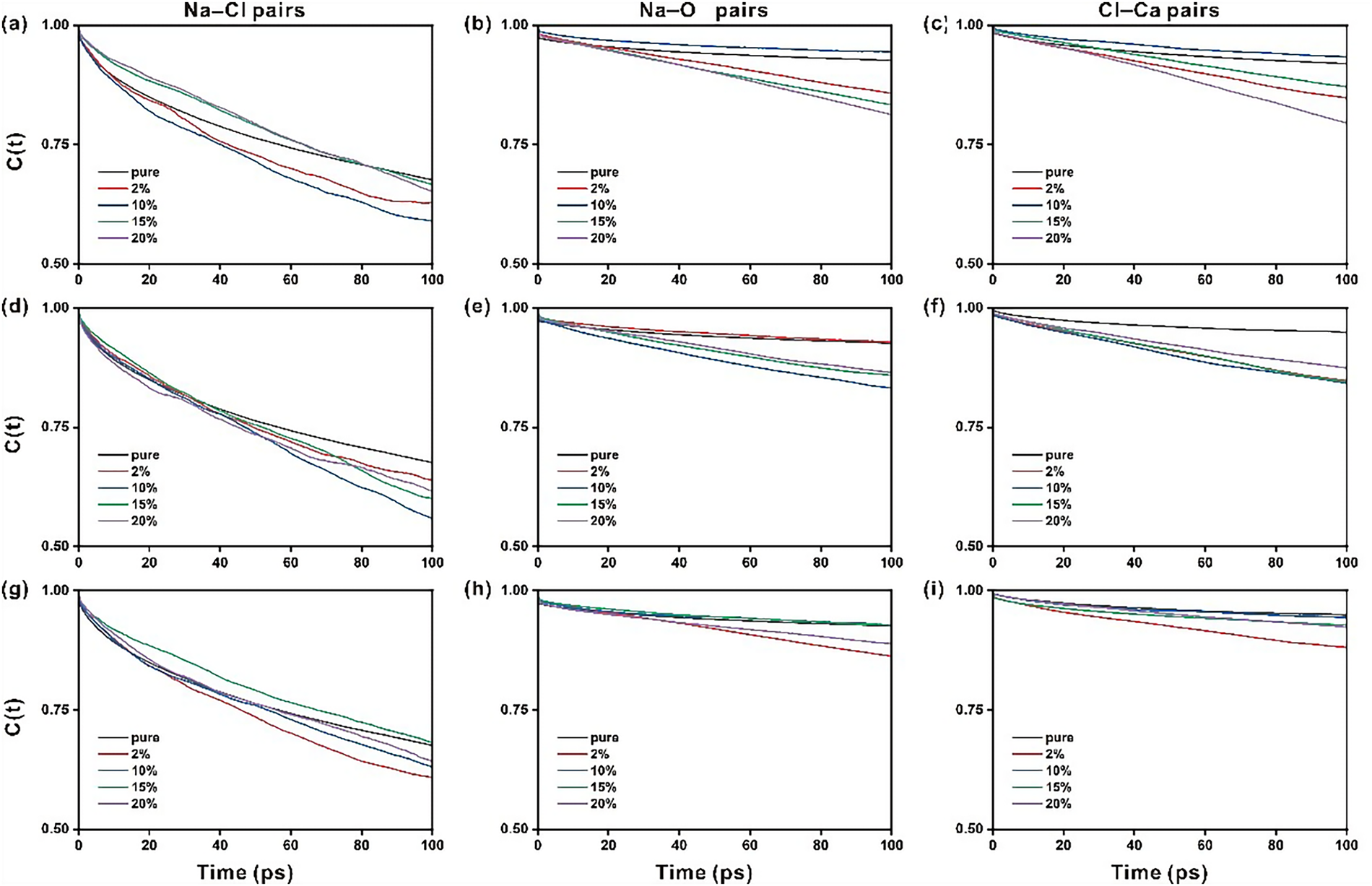
Figure 4 Time correlation function of Na-Cl, Na-O and Cl-Ca pairs on the nanopore of Sr2+ doped ettringite (a–c), Fe3+ doped ettringite (d–f) and IO3− doped ettringite (g–i)
For Fe3+ doped ettringite, the influence of elemental doping on the ionic pair’s interactions can be similar to that of Sr2+ doped ettringite (Figure 4d–f). Generally, the ionic pair interactions, including Na-Cl, Na-Oh, and Cl-Ca pairs, would be weakened by Fe3+ doping. Relatively, the stability of Na-Oh and Cl-Ca pairs is more stable than that of Na-Cl pairs. For IO3− doped ettringite (Figure 4g–i), the influence of elemental doping on the ionic pairs interactions is inconspicuous compared with that of Sr2+ and Fe3+ doped ettringites. The elemental doping degree also exhibits a weaker impact on the stability of the ionic pair interactions. Among the 4 doping levels, only 2% IO3− doping shows a relatively apparent effect to weaken the stability of Na-Oh and Cl-Ca pairs.
Among the three types of doped ettringites (Sr-, Fe-, and IO3-doped), the Sr2+-doped ettringite was systematically explored. Figure 5 shows the RDF and TCF results of Cl_Sr pairs in the nanopore of Sr2+ doped ettringite. Firstly, comparing with Figure 3c (the RDF of Cl_Ca pairs in the nanopore of Sr2+ doped ettringite), it can be found that the RDF peak strength of Cl_Sr pairs is far higher than that of Cl_Ca pairs. It indicates that the Cl_Sr interactions are stronger than those of Cl_Ca. Besides, with the doping degree increase, the ion pair interactions are weakened for both Cl_Ca and Cl_Sr pairs. According to Figure 5b, the doping degree exhibits a relatively apparent influence on the stability of Cl_Sr pairs. With the Sr2+ doping degree increase, the stability of Cl_Sr interaction pairs is also increased. To further analyze the ion distributions, the number of key ion pairs is explored, as shown in Table S1, the coordination number of these ion pairs decreases as the doping level increases.
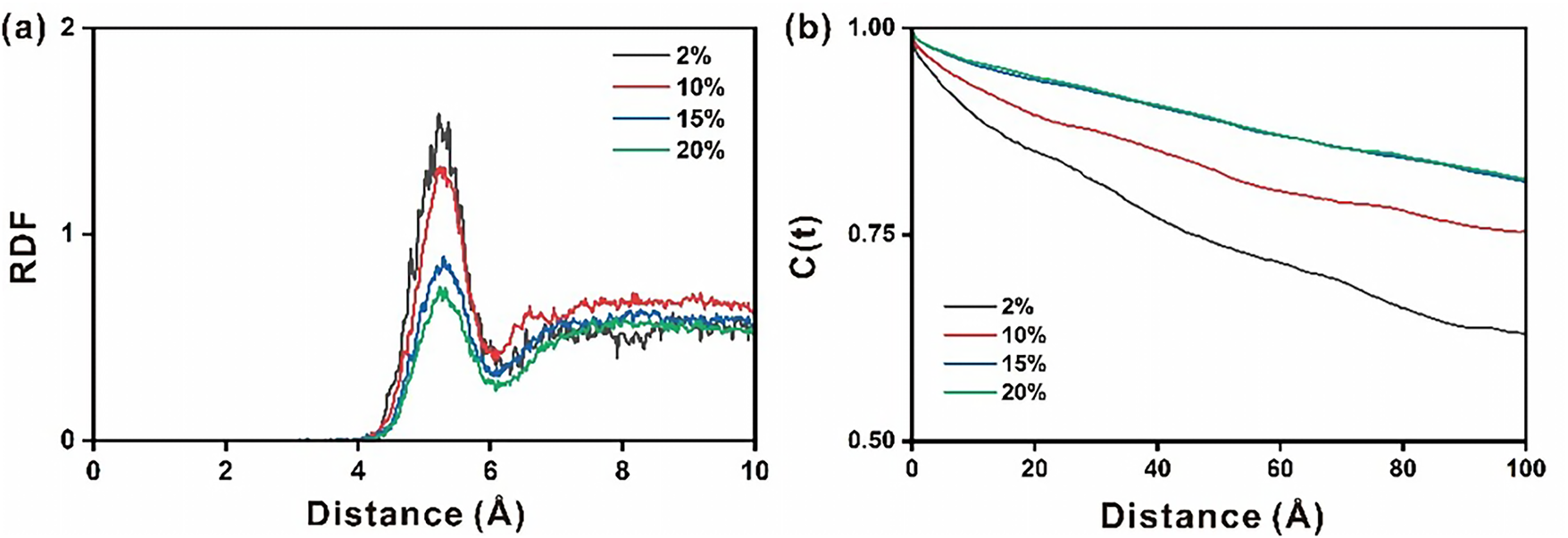
Figure 5 Radial distribution function (a) and time correlation function (b) of Cl_Sr pairs in the nanopore of Sr2+ doped ettringite
Further, the mean square displacement (MSD) analysis was also carried out to explore the ions dynamics behaviors in the nanopores of different doped ettringites (See Figure 6). For Sr2+ doped ettringite (see Table S2), the Na+ exhibits a stronger affinity for O atom of AlO4 tetrahedron and forms more stable ionic bonds, resulting in a significantly lower diffusion rate compared to Cl−. When the doping level is 10% or 15%, the MSD curves of Na+ are nearly identical, indicating that the diffusion coefficients at these concentrations are also very close. However, at a 20% doping level, the MSD curve of Na+ lies below those of other doping levels, suggesting that the diffusion process is inhibited, with a diffusion coefficient of 0.63 × 10−9 m2/s. For Cl−, at a 2% doping level, the MSD curve is notably lower than at other levels, indicating significant inhibition of diffusion, with a diffusion coefficient of 0.93 × 10−9 m2/s. The doping level significantly affects the diffusion behavior of both Cl− and Na+, while water molecules show minimal influence, as their MSD curves are almost overlapping across five doping levels, with nearly identical diffusion coefficients. The calculated diffusion coefficients are consistent with those reported for pristine ettringite [39].
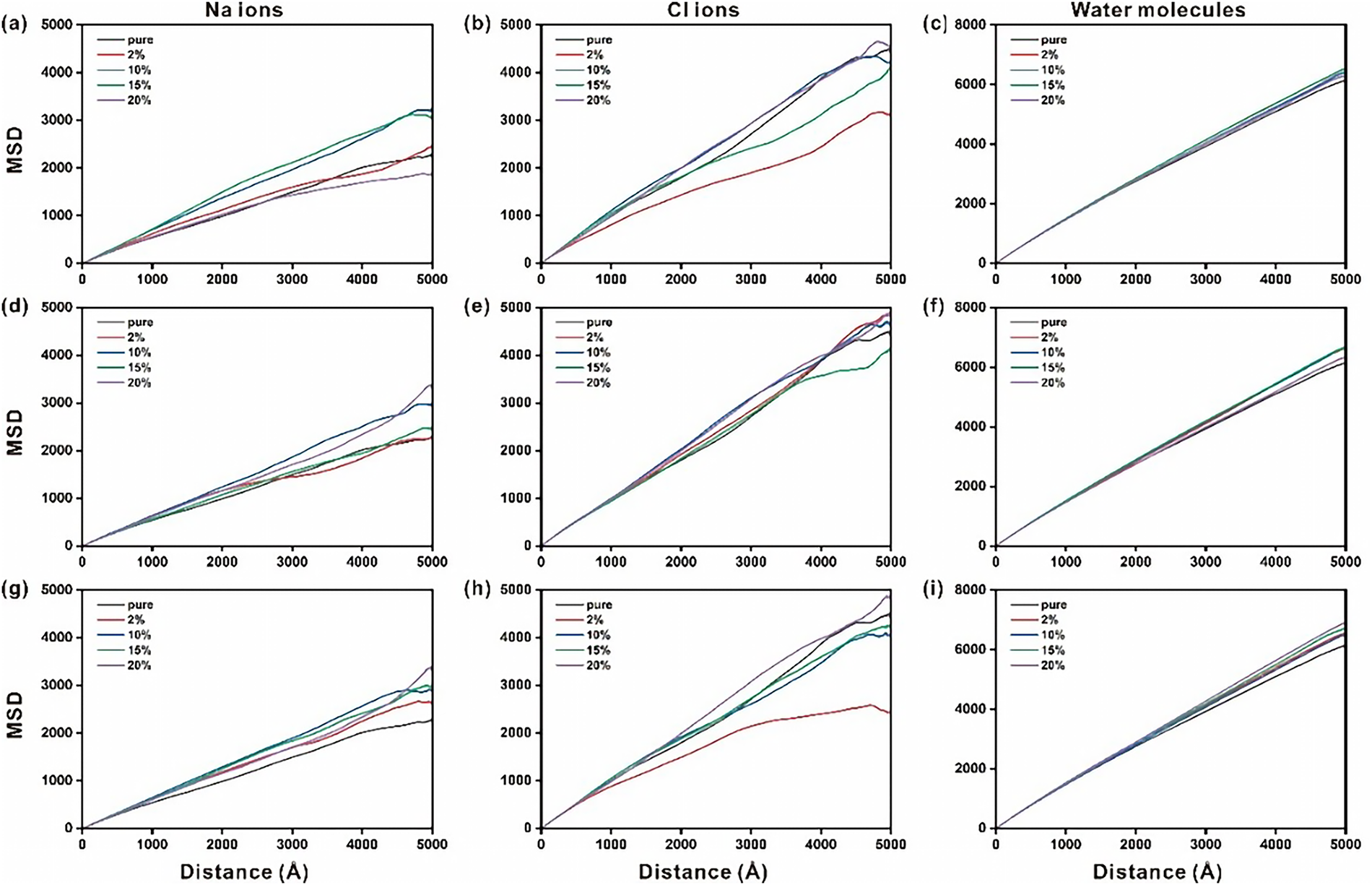
Figure 6 The mean square displacement of Na ions, Cl ions and H2O molecules on the nanopore of Sr2+ doped ettringite (a–c), Fe3+ doped ettringite (d–f) and IO3− doped ettringite (g–i)
For Fe3+ doped ettringite (see Fig. S1 and Table S3), the results of MSD indicate that changes in doping levels did not significantly affect the diffusion rates of sodium ions, chloride ions, and water molecules. Notably, during the first 2000 ps, the mean squared displacement (MSD) curves for Na+ and Cl− under different doping levels almost completely overlapped, suggesting that their diffusion behavior was largely unaffected by doping levels under these conditions. However, after 2000 ps, the MSD curves began to diverge, which may be attributed to changes in the adsorption stability of ions at the interface.
For IO3− doped ettringite (see Fig. S2 and Table S4), the analysis of the MSD curves of Na+, Cl−, and water molecules reveals distinct diffusion behaviors. The relationship among them is characterized as MSD(water) > MSD(Cl−) > MSD(Na+), indicating that water molecules diffuse the fastest, followed by chloride ions, and finally sodium ions. For Na+ diffusion, doping with IO3− increases the diffusion rate of Na+ compared to the pure base material. This suggests that the introduction of the IO3− enhances the mobility of Na+ ions through structural or electronic modifications in the host lattice. For Cl− diffusion, at a 2% doping level, the MSD curve of Cl− ions shows significantly lower values compared to other doping levels, indicating that Cl− diffusion is strongly suppressed under this condition. The calculated diffusion coefficient is 0.87 × 10−9 m2/s. This suppression is attributed to the direct interaction between Cl− and Ca2+ ions at 2% doping, which contrasts with the indirect interactions observed at other doping levels. A similar phenomenon has been reported: Na-doped tobermorite exhibits the hindered ability for Cl adsorption and diffusion [52].
3.3 The influence of ocean depth
Further, the impact of ocean depth, actually the hydrostatic pressure, on the stability and adsorption behavior of different ionic pairs in nanopores of the different elemental doped ettringites was also investigated. The TCF result of Sr2+ doped ettringite is shown in Fig. S3. It can be seen that in low doping concentrations, the ionic bond stability between Cl− and Ca2+ is superior to that between Cl− and Sr2+. As the doping concentration increases, the ionic bond stability between Cl− and Sr2+ gradually enhances, as the increased number of Sr2+ improves the coordination probability of Cl− with Sr2+. Under different doping concentrations, the effect of depth on the stability of Cl-Ca and Cl-Sr ion pairs differs significantly. For the Cl-Ca ion pair, increasing depth increases bond stability at doping concentrations of 2%, 15%, and 20%. However, at 10% doping concentration, increasing the depth to 10 km results in a significant weakening of bond stability, whereas increasing it to 20 km results in a noticeable enhancement of bond stability. This indicates that, even at the same doping concentration, changes in depth have differential effects on ionic bond stability. For the Cl-Sr ion pair, at 2% and 10% doping concentrations, bond stability is significantly affected by depth changes. However, at 15% and 20% doping concentrations, bond stability shows a less pronounced response to depth changes. Specifically, at a doping concentration of 15%, the TCF curves for the five depths are very close to each other, and even overlap in a certain period, indicating that the bond stability is similar across the five depths under this doping concentration.
The stability of Cl-Ca and Cl-Sr ion pairs is lower than that of Na-Oh ion pairs (see Figure 4), which is the primary reason for the greater adsorption capacity of sodium ions over chloride ions at the interface. The TCF result of Fe3+ doped ettringite is shown in Fig. S4, and the stability of Cl-Ca ionic bonds is influenced by depth and doping levels in a complex manner. At a 2% doping level, increasing depth reduces the stability of the Cl-Ca ionic bond. However, at 10% and 20% doping levels, increasing the depth enhances bond stability. Notably, at a 15% doping level, the stability of the Cl-Ca bond decreases significantly at 5 km, but increases at other depths studied. This indicates that even under the same doping level, changes in depth can significantly affect the stability of ionic bonds. Combined with the analysis of Na-Oh ionic pairs (see Figure 4), it is evident that the stability of Cl-Ca ionic bonds is lower than that of Na-O ionic bonds, which explains why the sodium ion has a stronger adsorption capacity at the interface compared to the Cl−. The TCF result of IO3− doped ettringite is shown in Fig. S5. When the doping concentration is 2%, the stability of the Cl-Ca ionic bond decreases as the depth increases to 3 km or 20 km, but increases when the depth reaches 5 km or 10 km. For doping concentrations of 10% and 15%, the effect of depth on bond stability is less pronounced. Additionally, at a doping concentration of 20%, the stability of the Cl-Ca bond significantly weakens when the depth reaches 10 km, but enhances at other depths. This indicates that even under the same doping concentration, the impact of depth variation on ionic bond stability is heterogeneous. Combined with the previous analysis of the Na-Oh ionic pair, it can be concluded that the Cl-Ca ionic bond is less stable than the Na-Oh ionic bond, which is the primary reason for the greater adsorption capacity of sodium ions over chlorine ions at the interface.
In this study, molecular dynamics simulations were conducted to investigate the influence of dopants on the ion distribution and adsorption behavior on the nanopore’s surface in ettringite (AFT). Further, the effect of the ion dosage and pressure was also considered to explore the ion/AFT interfacial interactions under a certain deep-sea environment. The following conclusions were drawn:
(1) The ion dosage of Na+ or Cl− exhibits the apparent effect on the ion distributions on doped AFTs. With increasing ion dosage, more ions come to and distribute around the salt solution/AFT interface, and there are more opportunities for ion pair formation.
(2) The surfaces of the three different doped ettringites can adsorb both cations and anions from the solution, but the adsorption mechanisms are different. The adsorption of Na+ is primarily achieved through the formation of a stable coordination with the O atom of the AlO4 tetrahedron on the surface. In contrast, the adsorption of Cl− mainly occurs through direct interaction with Ca2+ (such as Sr2+ in Sr-doped ettringite) on the surface, as well as through indirect interaction with the Na+ already adsorbed on the surface.
(3) The types of doped ions, doping concentration, and ocean depth significantly influence the stability of ion pairs in nanochannels in ettringite, thereby affecting ion diffusion. The variation in doping concentration may have a complex effect on the mobility of ions, potentially exhibiting either inhibitory or promoting effects. Additionally, increasing ocean depths tend to suppress ion mobility, and the mobility of ions remains relatively similar across the four depths considered, indicating that the inhibitory effect is not linearly related to the increase in depth.
Acknowledgement
The authors acknowledge the financial support from the National Natural Science Foundation of China, China Postdoctoral Science Foundation and the Key Development Projects of the Sichuan Provincial Science and Technology Plan.
Funding Statement
The authors acknowledge the financial support from the National Natural Science Foundation of China (No. 52178253), China Postdoctoral Science Foundation (GZC20231792, 2023M742463) and the Key Development Projects of the Sichuan Provincial Science and Technology Plan (2022YFG0135).
Author Contributions
Luohao Fan: Conceptualization, software development, validation, data processing, drafting, review and editing, visualization. Pengfei Tang: Structure editing, review and editing, visualization. Laibao Liu: Review and editing. Honggang Zhang: Investigation, review and editing. Shuchun Hu: Hardware. Wei Feng: Investigation, review and editing. Run Zhang: Software, review and editing. Qingyuan Wang: Supervision and guidance, review and editing. Hongping Zhang: Conceptualization, supervision and guidance, fundraising, review and editing, resource provision. All authors reviewed and approved the final version of the manuscript.
Availability of Data and Materials
The data presented in this study are available on request from the corresponding authors.
Ethics Approval
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Supplementary Materials
The supplementary material is available online at https://www.techscience.com/doi/10.32604/zkg.2026.075582/s1.
Abbreviations and Full Names
| Abbreviation | Full Name |
| MD | Molecular Dynamic |
| C-S-H | Calcium Silicate Hydrate |
| CH | Portlandite |
| C-A-S-H | Calcium Aluminate Silicate Hydrate |
| RDF | Radical Distribution Functions |
| TCF | Time Correlation Function |
| MSD | Mean Square Displacement |
| NPT | Constant Number Pressure Temperature Ensemble |
| AFT | Ettringite |
[1] Kuang Y, Zhang Y, Zhou B, Li C, Cao Y, Li L, et al. A review of renewable energy utilization in islands. Renew Sustain Energy Rev. 2016;59(2):504–13. doi:10.1016/j.rser.2016.01.014.
[2] Kobayashi M, Takahashi K, Kawabata Y. Physicochemical properties of the Portland cement-based mortar exposed to deep seafloor conditions at a depth of 1680 m. Cem Concr Res. 2021;142:106335. doi:10.1016/j.cemconres.2020.106335.
[3] Shaikh SF, Mazo-Mantilla HF, Qaiser N, Khan SM, Nassar JM, Geraldi NR, et al. Noninvasive featherlight wearable compliant marine skin: standalone multisensory system for deep-sea environmental monitoring. Small. 2019;15(10):1804385. doi:10.1002/smll.201804385.
[4] Takahashi K, Kawabata Y, Kobayashi M, Gotoh S, Nomura S, Kasaya T, et al. Action of hydraulic pressure on Portland cement mortars—current understanding and related progress of the first-ever in-situ deep sea tests at a 3515 m depth. J Adv Concr Technol. 2021;19(3):226–39. doi:10.3151/jact.19.226.
[5] Li LG, Chen XQ, Chu SH, Ouyang Y, Kwan AKH. Seawater cement paste: effects of seawater and roles of water film thickness and superplasticizer dosage. Constr Build Mater. 2019;229:116862. doi:10.1016/j.conbuildmat.2019.116862.
[6] Qu F, Li W, Dong W, Tam VWY, Yu T. Durability deterioration of concrete under marine environment from material to structure: a critical review. J Build Eng. 2021;35(1):102074. doi:10.1016/j.jobe.2020.102074.
[7] Kawabata Y, Takano D, Takahashi K, Iwanami M. In situ observation for the influence of hydraulic pressure on internal damage of cement-based materials. Mater Des. 2022;216:110556. doi:10.1016/j.matdes.2022.110556.
[8] Andrade C. Steel corrosion rates in concrete in contact to sea water. Cem Concr Res. 2023;165:107085. doi:10.1016/j.cemconres.2022.107085.
[9] Li G, Zhang A, Song Z, Shi C, Wang Y, Zhang J. Study on the resistance to seawater corrosion of the cementitious systems containing ordinary Portland cement or/and calcium aluminate cement. Constr Build Mater. 2017;157:852–9. doi:10.1016/j.conbuildmat.2017.09.175.
[10] Sun W, Mu R, Luo X, Miao C. Effect of chloride salt, freeze-thaw cycling and externally applied load on the performance of the concrete. Cem Concr Res. 2002;32(12):1859–64. doi:10.1016/S0008-8846(02)00769-X.
[11] Li H, Nie Q, Wang C, Wang G, Zhang L, Yuan L. Durability investigation of fractured coal-gasified ash slag concrete eroded by sulfate and chlorine salts. Case Stud Constr Mater. 2024;20:e02745. doi:10.1016/j.cscm.2023.e02745.
[12] Chen X, Zhang Q. A novel method to prevent chloride from accumulating on surface of reinforcement in concrete: embedding diaphragm. Constr Build Mater. 2024;411:134423. doi:10.1016/j.conbuildmat.2023.134423.
[13] Jin Z, Chang H, Du F, Zhao T, Jiang Y, Chen Y. Influence of SAP on the chloride penetration and corrosion behavior of steel bar in concrete. Corros Sci. 2020;171:108714. doi:10.1016/j.corsci.2020.108714.
[14] Chen J, Jia J, Zhu M. Development of admixtures on seawater sea sand concrete: a critical review on Concrete hardening, chloride ion penetration and steel corrosion. Constr Build Mater. 2024;411:134219. doi:10.1016/j.conbuildmat.2023.134219.
[15] Du X, Li G, Cao A, Wang A, Zhou Y. Study of transport and binding behavior of chloride ions in concrete under single-sided salt-freezing cycle. Constr Build Mater. 2025;471:140749. doi:10.1016/j.conbuildmat.2025.140749.
[16] Tian Y, Tian Z, Jin N, Jin X, Yu W. A multiphase numerical simulation of chloride ions diffusion in concrete using electron microprobe analysis for characterizing properties of ITZ. Constr Build Mater. 2018;178:432–44. doi:10.1016/j.conbuildmat.2018.05.047.
[17] Jain A, Gencturk B, Pirbazari M, Dawood M, Belarbi A, Sohail MG, et al. Influence of pH on chloride binding isotherms for cement paste and its components. Cem Concr Res. 2021;143:106378. doi:10.1016/j.cemconres.2021.106378.
[18] Ting MZY, Wong KS, Rahman ME, Meheron SJ. Deterioration of marine concrete exposed to wetting-drying action. J Clean Prod. 2021;278(1):123383. doi:10.1016/j.jclepro.2020.123383.
[19] He X, Zhou J. Mechanical characteristics of sea-sand concrete in simulated marine environment. Constr Build Mater. 2021;274(1):122098. doi:10.1016/j.conbuildmat.2020.122098.
[20] Hua T, Li Z, Hu X, Tang J, Zhang J, Liu G. Effects of CEA on the microstructure evolution and chloride ion migration of marine concrete. Constr Build Mater. 2022;358:129464. doi:10.1016/j.conbuildmat.2022.129464.
[21] Scrivener K, Ouzia A, Juilland P, Kunhi Mohamed A. Advances in understanding cement hydration mechanisms. Cem Concr Res. 2019;124:105823. doi:10.1016/j.cemconres.2019.105823.
[22] Zhao J, Shi J, Li Y, Hou P, Liang S, Chen H, et al. Phase and microstructure evolution of the hydration products of magnesium phosphate cements under sulfuric acid environments. Constr Build Mater. 2024;418:135465. doi:10.1016/j.conbuildmat.2024.135465.
[23] Zhutovsky S, Nayman S. Modeling of crack-healing by hydration products of residual cement in concrete. Constr Build Mater. 2022;340:127682. doi:10.1016/j.conbuildmat.2022.127682.
[24] He Z, Pei X, Zhang J, Huang R, Deng M, Gao Y, et al. Molecular simulation for the relationship between the functional groups of chemical admixtures and cement hydration product Ca(OH)2 in the grouting process. Ecol Indic. 2023;153:110404. doi:10.1016/j.ecolind.2023.110404.
[25] Gao Y, Luo J, Zhu X, Zhang J, Fan K, Ma M. A review on the effect of organic admixtures containing different functional groups on the hydration behaviors of Portland cement. Rev Inorg Chem. 2025. doi:10.1515/revic-2024-0014.
[26] Zheng Z, Li H, Hu Y, Luo S, Zhu J. Effect of nano-Ti-Li-Al hydrotalcite-like on the hydration and hardening properties of sulfoaluminate cement-based materials. Constr Build Mater. 2024;450:138607. doi:10.1016/j.conbuildmat.2024.138607.
[27] Kim N, Seo J, Lee HK. Enhancement in clinker hydration degrees and later stage-ettringite stability of calcium sulfoaluminate cements by the incorporation of dolomite. Cem Concr Compos. 2025;155:105815. doi:10.1016/j.cemconcomp.2024.105815.
[28] Chang J, Zeng T, Li J. Influence of iron substitution on the microstructure and properties of ettringite in calcium sulfoaluminate cement: a comprehensive study on the Al/(Fe+Al) ratios. J Build Eng. 2024;86:108855. doi:10.1016/j.jobe.2024.108855.
[29] Zhao Y, Sun Y, Guo Z, Qiu J, Sun X. First-principles investigations of arsenate doping into the ettringite lattice. J Clean Prod. 2023;419:138266. doi:10.1016/j.jclepro.2023.138266.
[30] Fan C, Wang B, Xu Y. Solidification/stabilization and immobilization mechanism of Pb(II) and Zn(II) in ettringite. Cem Concr Res. 2023;174:107350. doi:10.1016/j.cemconres.2023.107350.
[31] Zhong H, Yang L, Wang F. Properties of (Al, Fe)-ettringite solid solution: experiment, atomic simulation, and thermodynamics modeling. Cem Concr Res. 2024;182:107556. doi:10.1016/j.cemconres.2024.107556.
[32] Bouibes A, Laanaiya M, Lacarrière L. Microscopic effect of iron dosage on the stability of Fe-doped ettringite. J Am Ceram Soc. 2024;107(7):5127–38. doi:10.1111/jace.19811.
[33] Guo B, Xiong Y, Chen W, Saslow SA, Kozai N, Ohnuki T, et al. Spectroscopic and first-principles investigations of iodine species incorporation into ettringite: implications for iodine migration in cement waste forms. J Hazard Mater. 2020;389:121880. doi:10.1016/j.jhazmat.2019.121880.
[34] Tararushkin EV, Pisarev VV, Kalinichev AG. Atomistic modeling of the structural and dynamic properties of aqueous NaCl and Na2SO4 solutions in the interlayer space of ettringite. Russ J Phys Chem A. 2022;96(4):818–23. doi:10.1134/S0036024422040318.
[35] Qi C, Manzano H, Spagnoli D, Chen Q, Fourie A. Initial hydration process of calcium silicates in Portland cement: a comprehensive comparison from molecular dynamics simulations. Cem Concr Res. 2021;149:106576. doi:10.1016/j.cemconres.2021.106576.
[36] Tu Y, Wen R, Yu Q, Cao J, Ji Y, Sas G, et al. Molecular dynamics study on coupled ion transport in aluminum-doped cement-based materials. Constr Build Mater. 2021;295:123645. doi:10.1016/j.conbuildmat.2021.123645.
[37] Pei T, Sun D, Wang Y, Wang J, Cui S, Li H, et al. Molecular simulation of the structure and mechanical properties of Al(Fe)–ettringite. J Mater Sci. 2024;59(19):8298–317. doi:10.1007/s10853-024-09676-4.
[38] Izadifar M, Sekkal W, Dubyey L, Ukrainczyk N, Zaoui A, Koenders E. Theoretical studies of adsorption reactions of aluminosilicate aqueous species on graphene-based nanomaterials: implications for geopolymer binders. ACS Appl Nano Mater. 2023;6(18):16318–31. doi:10.1021/acsanm.3c02438.
[39] Tararushkin EV, Pisarev VV, Kalinichev AG. Atomistic simulations of ettringite and its aqueous interfaces: structure and properties revisited with the modified ClayFF force field. Cem Concr Res. 2022;156:106759. doi:10.1016/j.cemconres.2022.106759.
[40] Tararushkin EV, Pisarev VV, Kalinichev AG. Interaction of nitrite ions with hydrated portlandite surfaces: atomistic computer simulation study. Materials. 2023;16(14):5026. doi:10.3390/ma16145026.
[41] Hong SN, Yu CJ, Ri KC, Han JM, Ri BH. Molecular dynamics study of the effect of moisture and porosity on thermal conductivity of tobermorite 14 Å. Int J Therm Sci. 2021;159:106537. doi:10.1016/j.ijthermalsci.2020.106537.
[42] Meng J, Lyu C, Wang L, Wang J, Nie B, Lyu Y, et al. Effect of cyclic load on mechanical properties and failure mechanisms of different rank coals. Energy. 2023;278:127934. doi:10.1016/j.energy.2023.127934.
[43] Jaeger R, Vancso GJ. An ab initio and force field study on the conformation and chain flexibility of the dichlorophosphazene trimer. Macromol Theory Simul. 1996;5(4):673–89. doi:10.1002/mats.1996.040050404.
[44] Li X, Zhang H, Zhan H, Tang Y. Structural and mechanical properties of doped tobermorite. Nanomater. 2023;13(16):2279. doi:10.3390/nano13162279.
[45] Gillispie EC, Mergelsberg ST, Varga T, Webb SM, Avalos NM, Snyder MMV, et al. Competitive TcO4−, IO3−, and CrO42− incorporation into ettringite. Environ Sci Technol. 2021;55(2):1057–66. doi:10.1021/acs.est.0c06707.
[46] Meng Z, Zhang Y, Chen WK, Fu CQ, Xiong QX, Zhang CL, et al. A numerical study of moisture and ionic transport in unsaturated concrete by considering multi-ions coupling effect. Transp Porous Medium. 2024;151(2):339–66. doi:10.1007/s11242-023-02011-6.
[47] Liu QF, Easterbrook D, Yang J, Li LY. A three-phase, multi-component ionic transport model for simulation of chloride penetration in concrete. Eng Struct. 2015;86:122–33. doi:10.1016/j.engstruct.2014.12.043.
[48] Tong L-Y, Qing XX, Zhang M, Meng Z, Meftah F, Liu Q-F. Multi-scale modelling and statistical analysis of heterogeneous characteristics effect on chloride transport properties in concrete. Constr Build Mater. 2023;367(5):130096. doi:10.1016/j.conbuildmat.2022.130096.
[49] Zhang W, Hou D, Ma H. Multi-scale study water and ions transport in the cement-based materials: from molecular dynamics to random walk. Microporous Mesoporous Mater. 2021;325:111330. doi:10.1016/j.micromeso.2021.111330.
[50] Sekkal W, Izadifar M, Zaoui A, Ukrainczyk N, Koenders E. Theoretical investigation of protective graphene-coated metakaolin geopolymer interface under moisture and chemical composition effects. Powder Technol. 2023;430(1):119007. doi:10.1016/j.powtec.2023.119007.
[51] Balucani U, Howard Lee M, Tognetti V. Dynamical correlations. Phys Rep. 2003;373(6):409–92. doi:10.1016/S0370-1573(02)00430-1.
[52] Abdelkawy A, White CE, Youssef M. Molecular simulation of chloride ion binding mechanisms to Na-doped tobermorite 14 Å as a model system for sodium-containing cements. J Phys Chem C. 2023;127(36):17958–77. doi:10.1021/acs.jpcc.3c03010.